- Science Castle Research Fund
- Announcements for middle and high school students
- Information for Teachers
- Digest of Oral Presentations
- Research Tips
Bioinformatics Research Opens the Way to Future Medical Research [Digest of Science Castle 2023 Kanto Conference].
2024.05.31![Bioinformatics Research Paves the Way for Future Medical Research [Digest of Science Castle 2023 Kanto Conference]](https://s-castle.com/wp-content/uploads/sites/14/2024/05/17f4f5db429d3b0e899de93c31332334.jpg)
Here is a digest of the Science Castle, an academic conference for junior and senior high school students where the next generation of researchers is in full swing, as well as the excitement of the event. In this issue, we report on the presentation by Yonami Kakuno (2nd year student at Sanda Kokusai Gakuen High School), who won the Grand Prize for the oral presentation at the Science Castle 2023 Kanto Conference!
Affiliation and grade are as of the time of the announcement.
To convey the appeal of bioinformatics
Hello, my name is Yonami Kakuno. My name is Yonami Kakuno from Sanda International School High School. My favorite things are meat, cultured meat and protein. I usually watch anime and do research on meat production at the University of Tokyo.
My ambition is "to integrate the wisdom of mankind and develop science and technology. I heard that there will be a total of about 131 oral and poster presentations at this year's Science Castle, and it would be a waste to keep those 131 presentations independent, so I would like to capture them as big data and contribute to the development of science and technology. In Big Data, the use of biological information is called "bioinformatics. I started this research because I was fascinated by the fascination of bioinformatics. I will be happy if I can convey this fascination to you through this presentation.
Predicting disease onset from unknown amino acid mutations!
It is well known that our biological information is composed of DNA, which determines the sequence of amino acids through the processes of transcription and translation.
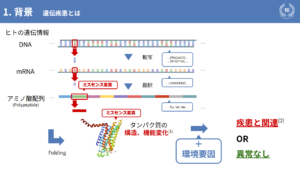
When missense mutations occur, it is known that the amino acid sequence is folded, i.e., the structure and function of the protein is altered. Here, lifestyle factors such as "drinking too much alcohol" or "smoking too many cigarettes," for example, can combine to cause our diseases or not.
Currently, missense mutations can be diagnosed by genetic testing. This is done by querying existing databases for mutations and making a diagnosis based on the data, but there is a problem: "We cannot deal with unknown amino acid mutations that are not known.
Therefore, the purpose of this study was set to explore the relationship between amino acid mutations and disease onset, and to predict disease onset from unknown amino acid mutations.
There are several similar studies that have been done from certain perspectives, such as predictors specific to protein conformational status, but none of them are complete and other views and further research are needed.

Study 1: Different proteins are associated with different diseases depending on where they function.
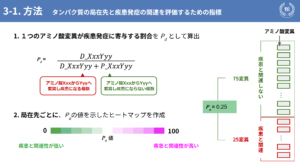
In this study, we used three main data sets to predict disease associations. The first hypothesis was that proteins may have different associations with disease depending on where they function (localization). In this study, we categorized proteins in the following subcellular organelles: nucleus, mitochondria, plasma membrane, cytoplasm, secretory, endoplasmic reticulum, and Golgi apparatus, and examined their association with disease, respectively.

Let me explain the index Pd that we have created in this study: Pd is "the ratio of the contribution of a single amino acid mutation to the development of a disease. For example, if a mutation of amino acids Xxx to Yyy occurs, it is calculated by dividing the number of mutations associated with the disease by the total number of mutations. Using this as an indicator, we visualized the disease association by creating a heat map showing the value of this Pd for each of the localization sites mentioned earlier.
The following are the results. Of the seven localization sites introduced earlier, two are shown here with particular differences: Fig. 2 shows the mitochondria and Fig. 3 shows the nucleus. Fig. 2 shows mitochondria and Fig. 3 shows nuclei, showing that mitochondria are highly associated with disease and nuclei are less associated with disease. Fig. 4 is a box-and-whisker diagram showing the distribution of Pd values for the seven localization sites and others shown earlier. As can be seen from here, one of the results of this study is that different localization sites have different associations with disease.
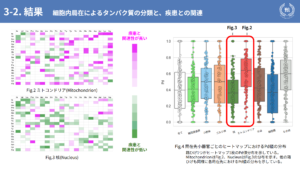
Mitochondria and nucleus, both of which were picked up earlier, are cell organelles that form the basis of biological activities, but proteins localized in mitochondria are not resistant to mutation, while those localized in the nucleus are considered to be resistant. One consideration is that although many transcription factors localize to the nucleus, they do not have a unique three-dimensional structure until they bind to DNA or RNA, making them less likely to undergo structural changes, one of the factors in the pathogenesis of disease, and thus may have acquired resistance.
Study 2: Building machine learning models that correctly predict disease onset
Then, two machine learning models were constructed to predict disease onset from unknown amino acid mutations using "conservation of amino acid sequence of protein," "localization information," "information on amino acid mutations," "information on biological changes," and "structural information," which is used in many models in previous studies. The one shown on the left is [Model 1: machine learning model without structural information], and the one on the right is [Model 2: machine learning model with structural information]. Table 2 and Table 4 show the accuracy of predicting disease onset from unknown amino acid mutations. Model 1 has an accuracy of approximately 82%, while Model 2 with structural information has an accuracy of approximately 81%, indicating that disease onset can be correctly predicted from unknown amino acid mutations.

What is surprising here is the unexpected result that the accuracy of both was the same in the model I constructed, even though previous studies have shown higher accuracy with the addition of structural information. As a consideration, the secondary structure of the protein is employed in this study, which is calculated from the amino acid sequence of the protein using an algorithm for estimating hydrogen bonds. However, the secondary structure cannot be calculated for proteins whose structures are not known, so the number of data was reduced, which may have caused a bias in the training data. Therefore, additional analysis is needed on this point, and we cannot make a definite conclusion at this point. We also compared the accuracy of our model with that of previous studies; Fig. 6 shows the ROC curve of our model and Fig. 7 shows the ROC curve of previous studies. The higher the number, the better the accuracy. The maximum accuracy of this study is about 0.9, while the maximum accuracy of the previous study is about 0.87. Although the training data is different, it was noted that the accuracy of this model is also comparable to that of the previous study.

Research for the sake of research in medicine, and then to clinical applications.
Finally, I would like to talk about future prospects. First, as a bioinformatics study, we would like to examine how subcellular localization is related to disease onset by subdividing the heat map we created this time, for example, we only used information on protein localization this time, but we would like to add information to multiple localization sites and compare the localization with their functions. We would like to add information to multiple localizations and compare them with the functions of the localizations.
I also believe that this research has two research significance: one is research for research's sake. For example, it is research for basic medical research or drug discovery research. The other is for clinical applications. For these two purposes, we would like to brush up our prediction model without using structural information. That is all. Thank you very much for your attention.

question and answer session
Yasuhiro Iida (Kanagawa Institute of Technology):
Thank you for your presentation. I would like to ask you a question. Are the locations of missense mutations considered random, or are they in the active site?
Yonami Kakuno:
Some are in the active site and some are not. However, when we analyzed the data against the active site, we found that not very many of them were in the active site.
Yasuhiro Iida (Kanagawa Institute of Technology):
Are you researching this with reference to clinical data?
Yonami Kakuno:
Yes, I am aware of this. The data is actually looking at the association between amino acid mutations and disease. We are using a database that has information on mutations and data on whether or not a disease has developed.
Tetsushi Nishiyama (Liverness Corporation):
I was wondering if there is any bias in the mutations registered in the database for each localization that you were talking about at the beginning. For example, in the case of data on mitochondria, I would like to ask whether there is any bias in the original data set, such as what is the percentage of diseases among the mitochondria data.
Yonami Kakuno:
Yes, thank you for your question. Certainly, the total number of entries for mitochondria is quite small, and if I may be more specific, the nucleus, plasma membrane, and cytoplasm have about 15,000 entries, and mitochondria have about 5,000 entries, so it is certainly possible that bias was introduced because of the difference in total number here. It is certainly possible that the total number of entries is different here, and thus bias may have occurred.
Tetsushi Nishiyama (Liverness Corporation):
That is something that needs to be thoroughly examined again.
Yonami Kakuno:
We would like to align the overall number by subdividing it more closely to account for bias.
Naoki Morishita (Nippon Ham Co., Ltd.):
Thank you for a very interesting presentation. Regarding the ultimate goal, since you talked about predicting unknown diseases, if we know the localization of the protein, or if we know what amino acids have been changed, we can predict how much the incidence of the disease will be. Am I correct in thinking that you would like to create such a model?
Yonami Kakuno:
Yes, that is correct. Thank you for your question. As I have written here, I would like to brush up my method of using subcellular localization information instead of structural information.
Kenjiro Hanaoka (Keio University):
I'm going to talk about a technical topic since the presentation was quite specialized, but I think that after the final prediction is made, it will have to be verified as a science.
Yonami Kakuno:
Yes, I think that's right. For example, if we predict the location of a mutation in this research, we need to verify whether a mutation in that location really causes a disease by conducting actual experiments using mice, etc. I think this will lead to the assistance of basic research and drug discovery research mentioned here. I think this would lead to the elucidation of the pathogenic mechanism of rare diseases, for example.
Kenjiro Hanaoka (Keio University):
If you go on to college or something and get the facilities to do such a thing, I thought it would be interesting to try it.
(*Honorifics omitted)